

Creating a scalable In Vitro Diagnostic (IVD) regulatory strategy is crucial for companies aiming to transition from startup to enterprise. This journey involves navigating complex regulatory landscapes, ensuring compliance, and maintaining flexibility to adapt to evolving requirements.
Also read: Regulatory Strategy, what is it and why do you need one?
Understanding IVD Regulatory Landscape Early
In Vitro Diagnostics (IVDs) are medical devices used to perform tests on samples taken from the human body, such as blood or tissue. These devices are essential for diagnosing diseases, monitoring health conditions, and guiding treatment decisions. The regulatory landscape for IVDs varies significantly across regions, with stringent requirements in place to ensure safety and efficacy.
In many cases with our IVDeology customers, the regulatory pathway is only considered part way through the development process. This is especially true for combination or companion diagnostics (CDx). The reality is, the earlier the path to compliance is understood and planned, the greater chance of success.
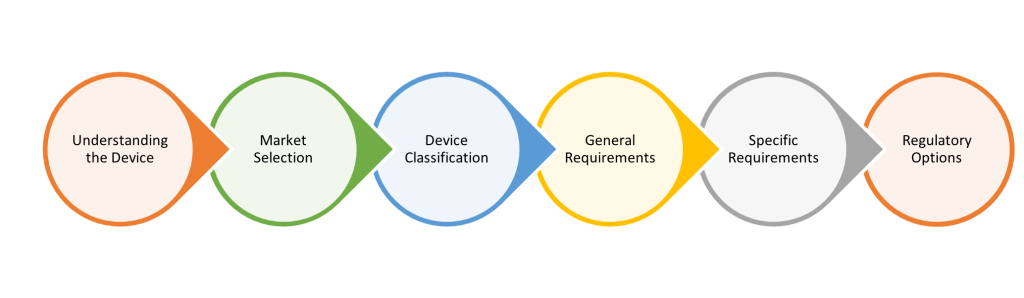
Figure 1 Considerations for building an effective regulatory strategy
Building a Scalable Regulatory Strategy
1. Early Engagement with Regulatory Authorities/Notified Bodies
Engaging with regulatory authorities and or Notified Bodies early in the development process is crucial. This helps in understanding specific requirements, obtaining guidance, and identifying potential challenges. Early engagement can also facilitate smoother approval processes and reduce time-to-market. For the US market, the FDA Q-Submission or Pre-Sub process, allows direct communication with the regulator to discuss your regulatory journey.
2. Risk-Based Classification
Understanding the risk classification of your IVD is fundamental. Different classes have varying requirements for clinical evidence, manufacturing practices, and post-market surveillance. Accurately classifying your IVD ensures that you meet the necessary regulatory standards without overburdening your resources.
In cases of a software as a medical device (SaMD) or IVD, you may have different components of the software which are IVDs, Medical Devices, or none of the above, so effective classification based on the intended purpose is key.
3. Robust Quality Management System (QMS)
Implementing a robust Quality Management System (QMS) is essential for ensuring compliance and scalability. A QMS should cover all aspects of the product lifecycle, from design and development to manufacturing and post-market activities. ISO 13485 is the internationally recognized standard for QMS in medical devices, including IVDs.
4. Clinical Evidence and Performance Evaluation
Generating clinical evidence and conducting performance evaluations are critical components of the regulatory strategy. This involves designing and executing clinical studies to demonstrate the safety and efficacy of the IVD. The data collected should be comprehensive and meet the requirements of the relevant regulatory jurisdictions.
5. Regulatory Submissions and Documentation
Preparing regulatory submissions and documentation is a meticulous process. This includes compiling technical documents, clinical study reports, risk management documents, and labeling information. Ensuring that all documentation is clear, organised, readily searchable and unambiguous is vital for successful approval.
6. Post-Market Surveillance and Vigilance
Post-market surveillance is an ongoing process that involves monitoring the performance of the IVD after it has been launched. This includes collecting and analyzing data on adverse events, product complaints, and other relevant information. Implementing a robust vigilance system helps in identifying and addressing issues promptly, ensuring continued compliance and patient safety.
Scaling from Startup to Enterprise
Building a scalable IVD regulatory strategy requires a comprehensive approach that encompasses early engagement with regulatory authorities, robust quality management systems, clinical evidence generation, meticulous documentation, and ongoing post-market surveillance.
By investing in scalable infrastructure, staying informed about regulatory changes, fostering cross-functional collaboration, and prioritizing training and development, companies can successfully transition from startup to enterprise while ensuring compliance and patient safety.
This journey is complex but achievable with the right strategy and commitment to excellence. As the IVD industry continues to evolve, staying proactive and adaptable will be key to long-term success.
Our approach
We believe that the regulatory strategy is the beginning of our customers journey in developing a rounded, and critical assessment of the regulatory landscape specific to the devices they are intending to place on the market. While in most case, the output in provided in a report, this should be considered a live document, with frequent review and update. This step is critical as the device performance, intended purpose or safety may change, and the regulatory environment may shift requiring a realignment of the strategy for regulatory success.
For further information contact [email protected]