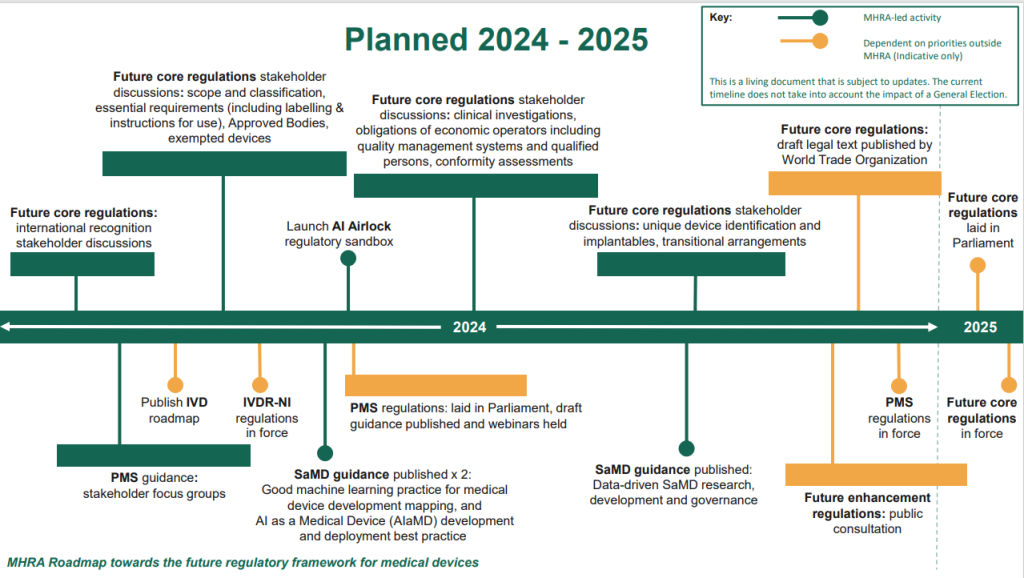
The MHRA have published a roadmap today which outlines the intended timelines for delivering the future regulatory framework for medical devices. Below is an Initial breakdown of the roadmap
There are 5 key areas of the roadmap, broken down into:
1- Implementation of future regulations
The breakdown ensures patients have access is to life changing devices and tests and introducing them safely and within regulatory compliance, in a way that also continues to showcase the UK as being attractive to medical innovators. It is intended that priority measures to enhance post-market surveillance will be put in place first in 2024, with core elements of the new framework expected to be in place in 2025.
2- Transitional agreements
It has introduced legislation which provides that CE marked medical devices may be placed on the Great Britain market to the following timelines:
- general medical devices compliant with the EU medical devices directive (EU MDD) or EU active implantable medical devices directive (EU AIMDD) with a valid declaration and CE marking can be placed on the Great Britain market up until the sooner of expiry of certificate or 30 June 2028
- in vitro diagnostic medical devices (IVDs) compliant with the EU in vitro diagnostic medical devices directive (EU IVDD) can be placed on the Great Britain market up until the sooner of expiry of certificate or 30 June 2030, and
- general medical devices, including custom-made devices, compliant with the EU medical devices regulation (EU MDR) and IVDs compliant with the EU in vitro diagnostic medical devices regulation (EU IVDR) can be placed on the Great Britain market up until the 30 June 2030.
This will enable certain CE marked medical devices to continue to be placed on the Great Britain market for longer.
The legislation provides that you can place self-declared CE marked Class I medical devices on the GB market beyond 30 June 2023 if they are:
- self-declared against EU MDR requirements (until 30 June 2030), or
- self-declared against MDD requirements before 26 May 2021 where Notified Body involvement in their assessment is not required under MDD but is under EU MDR (until 30 June 2028). This includes upclassified devices and reusable surgical instruments.
- It also provides that you can place a Class I medical device which has a sterile or measuring function with a valid MDD certificate on the GB market until 30 June 2028.
Class I medical devices and general IVDs under the Directives, for which the conformity assessment under the EU MDD or EU IVDD did not require a notified body, can only be placed on the Great Britain market if the involvement of a notified body would be required under the EU MDR or IVDR (i.e., if it is an upclassified device or a reusable surgical instrument Class I device). Custom-made devices that are compliant with the EU MDD or EU AIMDD can no longer be placed on the Great Britain market.
(Information above taken from GOV.UK website as of 9/1/2024)
3- Post Market surveillance requirements
The government have also introduced a list of requirements for manufacturers of devices for their post market surveillance once on the market (also known as PMS) in hopes to improve patient safety. The WTO website states that it intends to provide;
- Detail on what must be included as part of a PMS system, including the methods for collecting PMS data to support improved capturing of PMS data and harmonisation across manufacturers.
- Enhanced serious incident reporting obligations for manufacturers to support the detection of safety issues sooner.
- More stringent requirements for manufacturers to conduct periodic reviews of their PMS data, including for implantable medical devices. This aims to support manufacturers in earlier detection of trends/signals that may have an impact on the safety of a medical device.
4 – Future core regulations
During 2024, stakeholder discussion groups will be held to give early insight of the regulation policies of the future core regulations which will include:
– Ensuring devices have a unique device identification number (UDI)
– Improvements for implantable devices (class lll)
– Strengthening of QMS
– Changes and updates of class devices including those acting as software as a medical device (SaMD) as well as cyber security requirements and AI software in line with those in the EU
– New requirements for clinical investigations
– Requirements for a Qualified Person within regulatory internally in the company
– Requirements for intended purpose statements to align with their device
We will continue to monitor the updates as and when they are released, but any queries or questions about the above updates can be submitted to us via [email protected] or you can book time into our calendar and chat with our friendly team about how this will impact your business – get in touch here