

IVDeology have decades of IVD (invitro diagnostics) compliance support experience within the Quality and regulatory industry, with backgrounds from microbiology to lab based experience, and recently becoming part of the Abingdon Health PLC to offer a more holistic range of services from cradle to grave of diagnostic products. Co-founder and Director of training Nancy Consterdine talks about the US classifications of devices, having worked recently with US 510K submissions including pre-submissions.
The Global Harmonisation Task Force (GHTF) published their classification guidance in June 2006 proposing a risk based, 4 tier classification system for In Vitro Diagnostic Devices. This guidance has been widely used since 2006 for new regulatory systems being set up around the world e.g. the IVD Regulation in Europe 2017/746.
However, the United States of America (US) had already introduced a 3-tier risk-based classification system back in 1976 with the amendments to the Food, Drug and Cosmetics act (FD&C) to include medical devices. This system has now been in place for 48 years, despite amendments having been made around the regulatory pathways e.g. introduction of the 510K De Novo program, enacting of the Small Business Determination program and electronic submission processes. So, how does the classification system work currently and what are the future changes that the FDA are proposing?
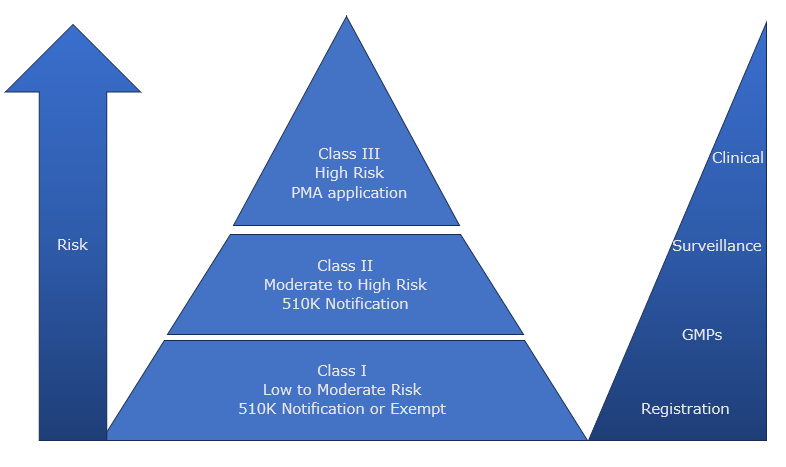
Class 1
- Low to Moderate Risk devices e.g. Transport culture medium, immunoelectrophoretic equipment, Biological stains
- Subject to General Controls (Regulatory Requirements which apply to all medical devices) all covered under a quality management system:
– Registration of producers of medical devices.
– Notifications and other remedies e.g. recall.
– Records and reports on devices e.g. adverse event report. - Manufacturers are still visible to FDA and may be subject to audit.
- Devices may be exempted from a General Control as stated in the classification regulation for that device e.g. they may be exempt from GMP other than keeping records and complaint files.
- Devices are submitted via the 510K pre-market notification process but some are exempt.
Class II
- Moderate to High Risk e.g. Blood Culture Assay, Rubella ELISA Test
- Subject to General Controls and Special Controls:
– Device specific
– Evidence of meeting performance standards
– Post market surveillance
– Adherence to guidelines
– Special labelling requirement - General Controls are considered insufficient to provide assurance of safety and effectiveness.
- Devices are submitted for pre-market notification via 510K process.
Class III
- High Risk e.g. Cancer Biomarker companion diagnostic assay
- Subject to General Controls and Premarket Approval:
– Quality Management processes and controls
– Software design, development and cyber security
– Analytical Verification data
– Clinical Performance Data - Pre-Market Approval (PMA) application is required
Recent Developments
In January 2024, a press release from Jeff Shuren, the director of the Centre for Devices and Radiological Health (CDRH) announced the intent to initiate the reclassification process for most IVDs which are currently class III (high risk) into class II (moderate risk). They identified that the majority of these tests are infectious disease and companion diagnostic IVDs. This is in line with the FDA least burdensome approach allowing manufacturers of some devices to seek marketing clearance through the 510K premarket notification route. In the release it also talked to the FDA desire to encourage more manufacturers to develop the test and in turn increase competition and access to these important tests.
The process of reclassification has already started with a panel meeting in September 2023 identifying 3 types of infectious disease diagnostic IVDs
- Nucleic acid and serology based IVDs to aid diagnosis of Hepatitis B Virus infection and management of infected patients.
- Serology based IVDs for detection of human parvovirus B19.
- Cell mediated immune reactivity IVDs to aid identification of in vitro responses to peptide antigens associated with TB infection.
Conclusion
The IVD industry can only welcome these moves by the FDA in conjunction with the amendment of the Quality Management System regulation (QMS) to be more closely aligned to the ISO 13485:2016 standard. It makes the USA a far more inviting prospect for initial market authorisation applications. The costs are transparent, the timelines are clearly identified and there is a process in place to present and discuss the device with the FDA in a pre-submission meeting.
There are however other considerations that need to be taken, Jeff Shuren has recently announced his retirement and Dr Michelle Tarver will assume the role of CDRH Acting Director. Also, there are presidential elections this year, will these changes impact the current trajectory of the CHRH? We at IVDeology will continue to monitor the situation across the pond, it is evident that since Brexit the UK government has been eager to foster recognition of other regulatory body approvals and it does feel that the movement of the FDA approval process is going in the same direction as that of the UK MDR.
Next steps to consider, and how we can help.
IVDeology Ltd can support with all of the above, please contact us for a friendly conversation to identify how we can support you with your compliance journey via our contact page