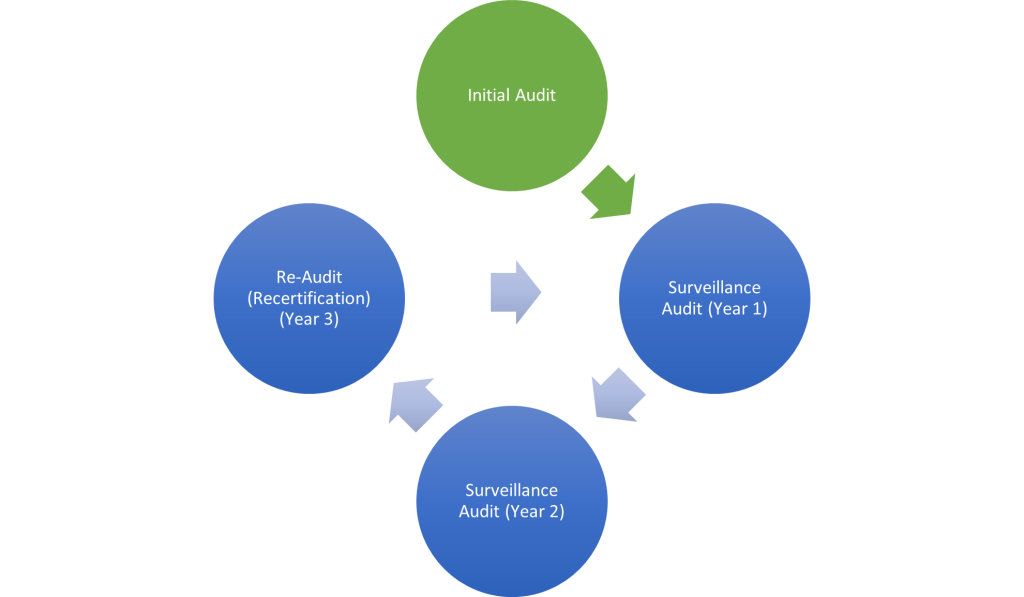
The first companion diagnostic (CDx) was approved by the US Food and Drug Administration (FDA) in 1998 with the European Medicines Agency (EMA) following with approval 2 years later focussed on the cancer treatment drug Herceptin. Since then, the rise of precision medicine has played an increasing part in eliminating the trial-and-error approach to identifying effective treatments for individual patients ensuring that therapies are tailored to their unique biological characteristics.
What Are Companion Diagnostics?
Companion diagnostics are in vitro diagnostic (IVD) tests (or may be an imaging tool), that provide critical information for the safe and effective use of a corresponding drug or biological product. These tests are co-developed with a therapeutic drug and are essential in determining whether a patient will benefit from a specific treatment, be at risk for severe side effects, or require adjustments in their therapy to achieve optimal results.
The labelling of a CDx must include the specific drug it was developed for and likewise the labelling of the drug must indicate the use of the CDx for prescription of the drug.
The US FDA approach to CDx Submission
In the US, the FDA is the central authority and requires that the drug and device are submitted for review and approval at the same time. Thus, the device and drug are developed at the same time and if possible, the device should be used as the Clinical Trial Assay (CTA). The therapeutic drug and it’s proposed CDx need Investigational Drug Application (IND) and Investigational Device Exemption (IDE) submissions and approvals before investigational clinical studies can commence.
During the development process the drug developer will be able to request meetings with the Centre for Drug Evaluation and Research (CDER) and the device developer will be able to meet with the Centre for Devices and Radiological Health (CDRH) via the Q-Submission process. For each of these meetings the respective developer should make it clear that they are co-developing the CDx and it is always useful to request a representative from the other centre to attend for awareness.
In the past the majority of CDx have been submitted and approved via the Pre-Market Approval process (PMA) and are classified as Class III devices. However, in 2024, the FDA announced plans to reclassify many Class IIIs to be Class II and enable manufacturers to submit for market authorisation via the 510K route.
The EU approach to CDx submission
In the EU, CDx are Class C following the classification rule 3(f) under the IVD Regulation 2017/746 and the key difference to the US process is the consultation process between the Notified Body of the device manufacturer and the EMA. The Notified Body will assess the technical documentation of the CDx and when that is complete, they will then consult with EMA for the final assessment alongside the drug file. This secondary process can add around 6 to 8 months to the approval process and needs to be planned carefully to allow sufficient time for market authorisations.
As with the US process, the CDx should be used as the CTA as early as possible, and the clinical use of the assay is often seen as an interventional clinical study requiring complex and lengthy applications to the separate member state competent authorities for approval.
The Future of Companion Diagnostics
Since the first CDx was approved by the FDA in 1998, there are now over 200 cleared or approved listed by the FDA. IVDeology Ltd is seeing the development of companion diagnostics growing alongside advancements in genomics and biotechnology. As our understanding of the genetic and molecular basis of diseases expands, manufacturers have the ability to create more precise and effective diagnostic tests, leading to better patient outcomes and more efficient healthcare systems.
Conclusion
Companion diagnostics are leading the way in the field of personalized medicine. By ensuring that treatments are tailored to the individual needs of patients, these diagnostics not only improve the efficacy and safety of therapies but also pave the way for a more personalized approach to healthcare. As technology continues to evolve, the role of companion diagnostics will undoubtedly become even more integral to modern medicine.
IVDeology Ltd can support small pharmaceutical developers understand the CDx requirements to enable clear and concise discussions with your identified device developers. Likewise we can support CDx developers understand the requirement for consultation with the FDA and the pathway to market approval.