The changeable regulatory environment for Medical Devices and In vitro Diagnostic medical devices (IVD) is showing no sign of slowing down as we begin 2025.
May 2025 – Certificates issued in accordance with IVD Directive (98/79/EC) Annex VI which shall become void at the latest on 27 May 2025 and can not be used for placing CE marked IVDs on the Great British Market.
June 2025 – Development of draft guidance on artificial intelligence (AI) development and deployment
Winter 2025 – Statutory Instrument on Pre-Market Requirements
With so much change happening, it has never been so important to engage with your regulatory teams, to understand the impact and implications of the changes and to get ahead of the game. IVDeology work with IVD manufacturers throughout the global industry to Educate, Evaluate and Execute regulatory strategies to Sustain market access.
You can get in touch with IVDeology by booking time with our friendly team here or email i[email protected] – we’d be happy to chat
We also have a series of online webinars that can also support your next steps and understanding of the regulatory landscape, you can check the early 2025 sessions here:
There has been a lot of discussion surrounding the focus of treatment within primary care. The NHS published guidance in August 2023 for integrating point of care IVD’s (NHS England » Integrating in vitro point of care diagnostics: guidance for urgent community response and virtual ward services). The Labour Party’s recent manifesto for the 2024 General Election stated that “Labour’s reforms will shift our NHS away from a model geared towards late diagnosis and treatment, to a model where more services are delivered in local communities” and “The National Health Service needs to move to a Neighbourhood Health Service, with more care delivered in local communities to spot problems earlier. To achieve this, we must over time shift resources to primary care and community services.” (Change Labour Party Manifesto 2024). Whilst this is good news for patients, it is not as simple as changing the location of the testing – there will be implications for the IVD industry in order to meet these proposals.
The In Vitro Diagnostic Medical Devices Regulation (Regulation (EU) 2017/746, ‘IVDR’) defines a “device for near-patient testing” as “any device that is not intended for self-testing but is intended to perform testing outside a laboratory environment, generally near to, or at the side of, the patient by a health professional.”
During Design & Development, IVD manufacturers have to determine the use environment and end users within their intended purpose. It is then their responsibility to demonstrate that the device is safe and effective when used in these environments by these intended end users and therefore there will be a number of additional requirements that need to be considered for devices intended for near-patient testing.
Performance Requirements:
The performance testing conducted will need to demonstrate that effective and reliable testing outside of the traditional controlled laboratory environments. Considerations such as the following will need to be considered:
Use Environment Testing:
Annex I section 9.4 states “The characteristics and performances of the device shall be specifically checked in the event that they may be affected when the device is used for the intended use under normal conditions:
(b) for devices for near-patient testing, performances obtained in relevant environments (for example, patient home, emergency units, ambulances).”
Testing of the device will therefore need to be conducted in the intended use environments. Manufacturers will need to show that the test can be used reliably in the environments that they indicate that the test can be used in. This could be, for example, doctor’s surgeries, A&E departments, patient’s homes or in ambulances. Any conditions specific for these environments would need to be considered e.g. vibrations or temperature fluctuations for devices intended to be used on an ambulance.
Usability Studies:
Annex I section 19.1 states that “Devices intended for self-testing or near-patient testing shall be designed and manufactured in such a way that they performappropriately for their intended purpose taking into account the skills and the means available to the intended user and the influence resulting from variation that can be reasonably anticipated in the intended user’s technique and environment. The information and instructions provided by the manufacturer shall be easy for the intended user to understand and apply in order to correctly interpret the result provided by the device and to avoid misleading information. In the case of near-patient testing, the information and the instructions provided by the manufacturer shall make clear the level of training, qualifications and/or experience required by the user.
It is important for manufacturers to have designed their device in such a way as to ensure that the intended end users can successfully use the device. Therefore it is vital that manufacturers conduct usability studies with their target end users who are often not laboratory trained personnel. This can then be used to demonstrate that consistent results can be obtained by these target end users, determine if the instructions provided with the device are adequate and help to identify if any training is required for the end users before the device can be used reliably.
Labelling Requirements:
The IVDR has also introduced specific labelling requirements for NPT (near patient testing) devices, including:
Devices labelled as Near-Patient Testing: The device label must indicate that the device is for near-patient testing. Although there are currently no symbols for this within ISO 15223-1:2021 Medical devices – Symbols to be used with information to be supplied by the manufacturer, MedTech Europe has provided some suggested symbols that can be used to indicate near-patient testing (New IVD symbols for compliance with the IVDR – MedTech Europe).
Individual Instructions for Use: Each individual device must be accompanied by its own instructions for use (IFU). For devices for professional use within a laboratory setting, if multiple devices were supplied then a single copy of the IFU could be provided if agreed by the purchaser. This is not allowed for devices intended for near-patient testing.
Paper-Based Instructions: According to Annex I 20.1(f) of the IVDR, the instructions for use must be provided in a physical paper format for near-patient tests, and, unlike laboratory based professional use devices, cannot be provided in electronic format.
Language Requirements: The languages that the Member States require for the device label and instructions for use for near-patient testing may be different to those for professional use only tests. This may add more translation costs on to manufacturers to access different markets.
The labelling provided must be appropriate to the device, its intended use, and the technical knowledge, experience, education, or training of the intended users. This will need to be considered by manufacturers when designing the labelling and tested during usability studies.
Notified Body Assessment
Whilst devices for near-patient testing are classified in their own right according to Annex VIII rule 4(b), the notified body assessment is slightly different. For Class B & Class C devices, the technical documentation of all devices for near-patient testing has to be assessed rather than the notified bodies sampling one technical file per generic device group or device category. Where a manufacturer has a number of devices for near-patient testing the increase of upfront cost to have their devices assessed will need to be considered.
Final thoughts
Whilst near-patient testing seems like a real win for patients and the direction of travel that the diagnostic industry is heading, this does provide some challenges for manufacturers. The additional burden to demonstrate that the device is effective and reliable when used outside of a laboratory setting and the potential increased upfront costs of conformity assessment is something that needs to be considered before being able to place the device on the market. For Great Britain, although the UK MDR 2002 does not specifically call out devices for near-patient testing currently, the indications from the MHRA on the future regulations is that it will be similar to the IVD Regulation with the new Essential Requirements being based on the General Safety & Performance Requirements. This is likely therefore to mean that the additional requirements for these types of devices will also be required here. However, if done correctly, near-patient testing will enable quicker diagnoses for patients and hopefully therefore better patient outcomes, which is ultimately what the IVD industry wants to support.
If you’d like to discuss near-patient testing or any of the compliance services that come along with it, from design and development to regulatory services, you can speak to us by dropping an email to [email protected] or book time with us via this link for when best suits you
The Medical Device Single Audit Programme (MDSAP) is a system by which the participant competent authorities to recognise the quality management certification (as awarded after audit against both ISO 13485 and county specific requirements) from a single authority for medical device and in-vitro diagnostic medical device (IVDs) legal manufacturers.
The programme has been established by the International Medical Device Regulators Forum (IMDRF) and is intended to provide a harmonised approach to demonstrating the compliance of the Quality Management System (QMS) using a globally recognised approach.
The MDSAP was developed by the IMDRF to:
Enable appropriate regulatory oversight of medical device manufacturers’ quality management systems while minimizing regulatory burden on industry;
Promote more efficient and flexible use of regulatory resources through work sharing and mutual acceptance among regulators while respecting the sovereignty of each authority;
Promote globally, in the longer term, a greater alignment of regulatory approaches and technical requirements based on international standards and best practices;
Promote consistency, predictability and transparency of regulatory programs by standardizing;
1. the practices and procedures of participating regulators for the oversight of third party auditing organizations, and 2. the practices and procedures of participating third party auditing organizations
Regulatory Authorities
MDSAP consists of Regulatory Authority Council Members, Observers and Affiliate members:
Regulatory Authority Council Members:
Therapeutic Goods Administration of Australia
Brazil’s Agência Nacional de Vigilância Sanitária
Health Canada
Japan’s Ministry of Health, Labour and Welfare, and the Japanese Pharmaceuticals and Medical Devices Agency
U.S. Food and Drug Administration
The RAC is the decision-making body of MDSAP and consists of representatives from all regulatory authorities that are members of the RAC. The RAC provides direction, oversight, and resources to support the MDSAP development, implementation, maintenance, and expansion.
Observer Members:
European Union (EU)
Singapore’s Health Sciences Authority (HSA) (NEW)
United Kingdom’s Medicines and Healthcare products Regulatory Agency (MHRA)
The World Health Organization (WHO) Prequalification of In Vitro Diagnostics (IVDs) Programme
The observers do not observe RAC members and do not attend RAC meetings, but they do observe and contribute the RAC activities. Both the EU and UK have been Observers for over 2 years, and as such, can apply to become full RAC members if desired.
Affiliate Members:
Argentina’s National Administration of Drugs, Foods and Medical Devices (ANMAT)
Ministry of Health of Israel
Kenya’s Pharmacy and Poisons Board (New member)
Republic of Korea’s Ministry of Food and Drug Safety
Federal Commission for Protection from Sanitary Risks (COFEPRIS) of Mexico
TFDA – Taiwan Food and Drug Administration
Affiliate members are not members of the RAC or an Official Observer, but engages in MDSAP, demonstrates understanding of MDSAP and utilizes MDSAP audit reports and MDSAP certificates for evaluating compliance with applicable medical device requirements, including a manufacturer’s quality management system, under the Affiliate Member’s regulatory framework.
The application of the MDSAP Programme
The utilisation of the MDSAP programme, and the resulting certificates are utilised differently depending on each Competent Authority as dedicated by each regional requirement.
Regulatory Authority
Utilisation of MDSAP
Australia
MDSAP audit report is used as part of the evidence that it has assessed for compliance with medical device market authorization requirements, unless excluded or exempt from these requirements.
Brazil
ANVISA utilizes the outcomes of the program as part of the pre-market and post market assessment.
Canada
Manufacturers intending to place a product on the market in Canada must have an MDSAP Certification issued by an Auditing Organization.
Japan
The Ministry of Health, Labour and Welfare (MHLW) and Pharmaceutical and Medical Devices Agency (PMDA) utilize these audit reports in pre-market and post-market audits.
United States
U.S. FDA will accept the MDSAP audit reports as a substitute for FDA routine inspections under a 510(k) device application. The use of MDSAP is not utilised for pre-approval or post-approval inspections for Pre-Market Approval (PMA) applications.
The regulations for the above Regulatory Authorities are available (English) from the USA FDA Website.
The use of the MDSAP programme and certification will be greater utilised as he programme expands and more regulatory authorities recognise the value in this process.
The MDSAP Audit Cycle and Auditing Organisations
Auditing Organisation are certification bodies that have successfully applied, and been recognised by the MDSAP programme to audit medical device manufacturers against the requirements of the MDSAP programme. The current list includes many European Notified Bodies (under EU IVDR), and UK Approved Bodies (under UKCA) and are globally recognised.
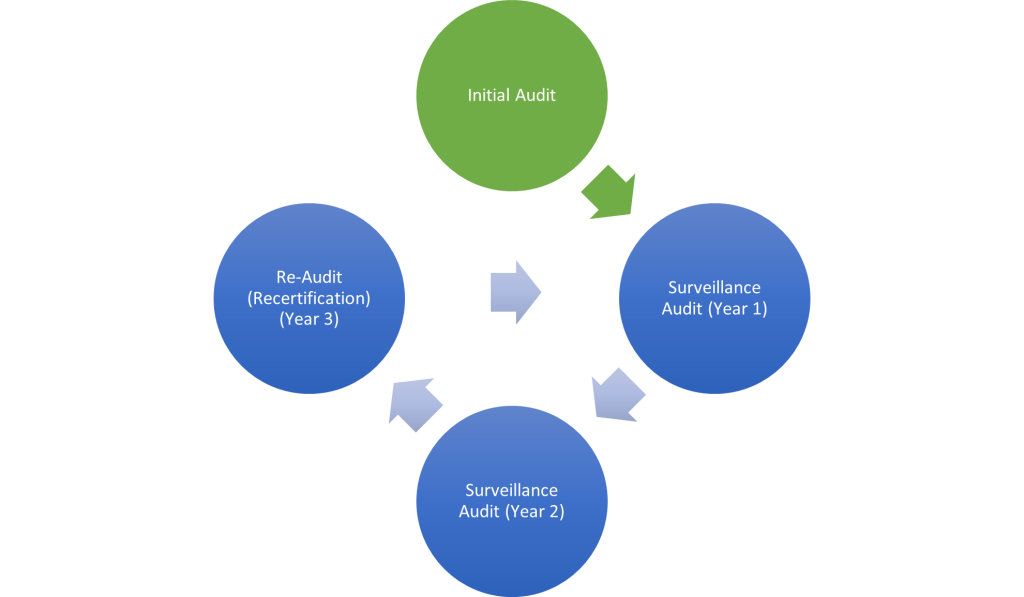
The MDSAP audit is typically build on to the existing ISO 13485 certification audit of the Quality Management System with a 3-year audit cycle.
Figure 1 MDSAP Certification Cycle
Will MDSAP come to the EU and UK?
There is much talk regarding the use of MDSAP by the EU and UK. While I am a strong advocate in global harmonisation, the existing members joined MDSAP to find a harmonised way to create a robust process for standardising QMS requirements from a position where they needed to find a suitable and robust process. Regardless of the state of play in the EU and UK, both regions already have robust mechanisms for the surveillance of ISO 13485, largely provided by the technical and commercial expertise of EU and UK Notified Bodies and Approved Bodies (as part of ISO13485 certification/IVDR conformity assessment), so the utilising MDSAP would be less impactful.
The opportunity would be regarding the outward facing regulatory convergence of EU and UK to align, and reduce burden for accessing other markets, MDSAP would be a good way of doing this. We have seen TGA become well placed in MDSAP/IMDRF mainly utilising CE marking for supporting AUS market access.
The challenge with MDSAP is that each jurisdiction has specific requirements, which make the MDSAP process clunky. The key to an improved model is to remove local requirements as much as possible, however this is dependent on global alignment at a political as well as regulatory level.
If you’d like to discuss MDSAP with us, you can book a call with the IVDeology team as we navigate this new programme and what it means for you as a manufacturer or provider of IVD’s and devices with each of the individual requirements. Being part of BIVDA (British In Vitro Diagnostics association), we’re in a great position to be able to receive and understand information and distribute to our networks as it comes, in a reliable and digestible way.
Or if you’d like support in other areas of Quality assurance or regulatory compliance, we’d be happy to chat with you. We can support with Quality management system implementation, transfer or uplifting.
We’ll be keeping you up to date with MDSAP news on our LinkedIn page and website, so do follow up on our socials and keep up to date with IVDeology along with Abingdon Health PLC.
Written by Stuart Angell, MD and Co-founder of IVDeology and IVDeology UKRP
Abingdon Health and IVDeology Ltd have decades of In-Vitro Diagnostic (IVD) regulatory compliance experience where our teams support your entire product journey from ‘cradle-to-grave’ to ensure you are getting your product to market in the quickest timeframe possible, reducing cost and strain, and keeping it compliant through its lifetime.
Abingdon Health also offer full-service contract development and manufacturing for lateral flow assays, bringing your idea to commercial success, with the benefit of an integrated regulatory and quality approach.
In our latest blog, Candice Vendettuoli Head of RAQA at Abingdon Health covers the importance of getting your Performance Evaluation (PE) right to streamline your route to market, and ensuring compliance to keep it there.
What is Performance Evaluation?
The In Vitro Diagnostic Regulation (IVDR) 2017/746, which entered into force in May 2017 and applied in May 2022, has introduced significant changes to the way in vitro diagnostic (IVD) medical devices are regulated in the European Union. One of the key aspects of this regulation are the new requirements for documenting the performance evaluation of IVD medical devices using a prescriptive document structure mandated within the Regulation.
Performance evaluation under IVDR is expected to be a continuous process throughout its entire lifecycle of the device. This process is crucial for ensuring that the device meets upon entry to the market, and continues to meet, the intended clinical benefits and safety as claimed by the manufacturer.
The mandated documents should be written to provide a comprehensive and structured narrative for the reviewer giving a clear and logical explanation of how the device was developed, verified and validated against the intended use/purpose claimed by the manufacturer. These documents are a requirement of the Technial Documentation described in Annex II and forms an essential part of the submission to the Notified Body
The mandated documents, unless they can justify why such studies are not applicable are as follows:
Performance Evaluation Plan
Ideally written during the early development of the device and updated regularly, this document has content prescribed within Annex XIII section 1.1 of the Regulation. Manufacturers are required to establish and regularly update the performance evaluation plan that outlines the device’s characteristics and performance, as well as the process and criteria used to generate the necessary clinical evidence.
Scientific Validity
The concept of scientific validity under the In Vitro Diagnostic Regulation (IVDR) 2017/746 is a cornerstone in the performance evaluation of in vitro diagnostic (IVD) medical devices. It refers to the association of an analyte with a clinical condition or physiological state, which must be substantiated with a medical-scientific rationale evidenced through a systematic literature search
Analytical Performance
Analytical performance refers to a device’s ability to accurately and reproducibly measure an analyte, marker, or molecule, which is a strictly technical performance without the need for correlation with a targeted pathology. There are analytical performance characteristics mandated within Annex I section 9.1(a) including assessing the accuracy, sensitivity and specificity,
Clinical Performance
Clinical performance is defined as the ability of a device to yield results that are correlated with a particular clinical condition, physiological or pathological process, or target population and intended user. Manufacturers must demonstrate clinical performance through one or more of the following:
Clinical performance studies, carried out according to the IVDR requirements on clinical performance studies described in Articles 57-77, Annex XIII section 2 and, if applicable, Annex XIV for studies other than those using leftover samples
Scientific peer-reviewed literature on the device under evaluation, or
Published routine diagnostic testing.
Performance Evaluation Report
The report (also known as a ‘PER’) provides a summary of the clinical evidence collected through the previous reports. An assessment can then be made against the current state of the art in diagnostics and medicine that a positive benefit-risk ratio of using the device for its intended purpose has been met and then all data has been collected.
This rigorous approach ensures the reliability and effectiveness of in vitro diagnostic devices within the European Union, with the primary aim of protecting public health by requiring high levels of safety and performance of these devices to be evidenced.
For manufacturers, understanding and adhering to the IVDR’s performance evaluation requirements is vital for successful market introduction of their IVDs in the European Market. It involves a comprehensive understanding of the general safety and performance requirements (GSPR), as well as the specific guidelines on performance evaluation stipulated in Article 56 of the IVDR.
Abingdon Health and through its subsidiary IVDeology Ltd , can offer support and guidance to help companies navigate these new and complex EU Performance Evaluation requirements.
We offer a full-service solution for all your regulatory and quality requirements including:
Person Responsible for Regulatory Compliance (PRRC)
Contact Us today to book some time with one of our industry experts to understand how we can support you bringing your product to market and keeping it there.