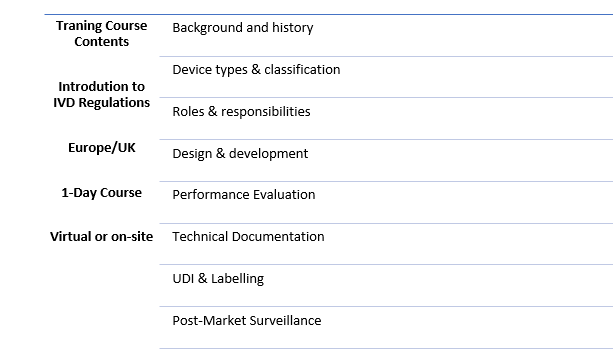
As part of the first budget prepared by the new UK Labour Government, a new Life Sciences Innovative Manufacturing Fund was announced.
“The government has committed up to £520 million for life sciences manufacturing, to help deliver on the government’s missions to kickstart economic growth and build an NHS fit for the future.
The LSIMF will be UK and sector wide and will provide capital grants for investments in the manufacture of:
- Human medicines (this includes both the manufacture of active pharmaceutical ingredients (API) / drug substance and finished product / drug product).
- Medical diagnostics – for both disease identification and monitoring.
- MedTech products – all types of medical devices related to human health.”
This new initiative could offer an opportunity to grow an SME diagnostic company to enhance their manufacturing capabilities.
“To be eligible for the fund, your project must:
- Have a total cost (capital and non-capital costs) of at least £8 million
- Be located in the UK
- Be primarily a capital investment
- Be a single company investment (as opposed to forming a partnership between companies or other types of organisations)
- Require only the amount of grant requested to proceed. For example, without the specific amount of funding you are requesting your project wouldn’t go ahead or go ahead at a smaller scale, go ahead overseas or would be significantly delayed (3 years or more)
- Be a manufacturing project for the manufacture of:
- Human medicines (this includes both the manufacture of active pharmaceutical ingredients (API) / drug substance and finished product / drug product).
- Medical diagnostics – for both disease identification and monitoring
- MedTech products – all types of medical devices related to human health.
The fund is open to applications for both MHRA-licenced products and products in development where a MHRA licence is intended to be sought for commercial scale-up, for example a manufacturing project for clinical trials.
- Manufacturing facilities are required to work to Good Manufacturing Practice (GMP) and the facility be intended to support clinical and/or commercial manufacture of API or drug product.
- Manufacturers of medicinal diagnostics and medical devices must confirm that their device meets or intends to meet the requirements of the Medical Devices Regulations 2002.”
Source: Life Sciences Innovative Manufacturing Fund (LSIMF): application guide – GOV.UK
The reference to the requirement to meet, or plan to meet the Medical Device Regulations 2002. Currently this would require device manufacturers to understand and comply with the current UK regulations, and potentially any new updates relating to UKCA. It would be interested to learn what level of regulatory understanding the manufacturers are required to have and demonstrate as part of the application.
For ideas on building an effective regulatory strategy, find out more in our recent blog: ‘Regulatory Strategy: What is it, and why do I need one?’
You can reach out to us at [email protected] or you can book straight into our diary at a time suitable for you here